Quality, Not Speed: Building a Production Evaluation Framework for AI-Assisted Medical Document Authoring
An engineering case study from fourteen months at a mid-sized pharma company.

Introduction
Pharmaceutical medical information teams answer questions from healthcare professionals. A physician calls or writes in with a clinical query about a marketed drug, and the company's Medical Information group composes a written response grounded in approved literature and internal data. These response documents fall into two categories: Custom Response Documents (CRDs), drafted for a single inquiry, and Standard Response Documents (SRDs), maintained as reusable answers to recurring questions. Both are reviewed by a Medical Review Committee (MRC) before they leave the company. The MRC reviews for medical accuracy, regulatory compliance, citation integrity, and tone; until it approves, no document is sent.
The work has a bottleneck that is not what most people assume. It is not the writing. The writing is straightforward for a clinically trained medical writer who has the source material in front of them. The bottleneck is the source material itself: locating the relevant peer-reviewed studies, identifying the passages that bear on the question, grounding every claim in the document back to a specific paragraph in a specific approved source, and reconstructing the citation anchors in the document-management system so the MRC can audit the chain of evidence. Across a portfolio of 250 documents per year, the median document required 6-9 hours of active authoring time before our intervention, and the median MRC review took 15 days across 3 rounds of revision.
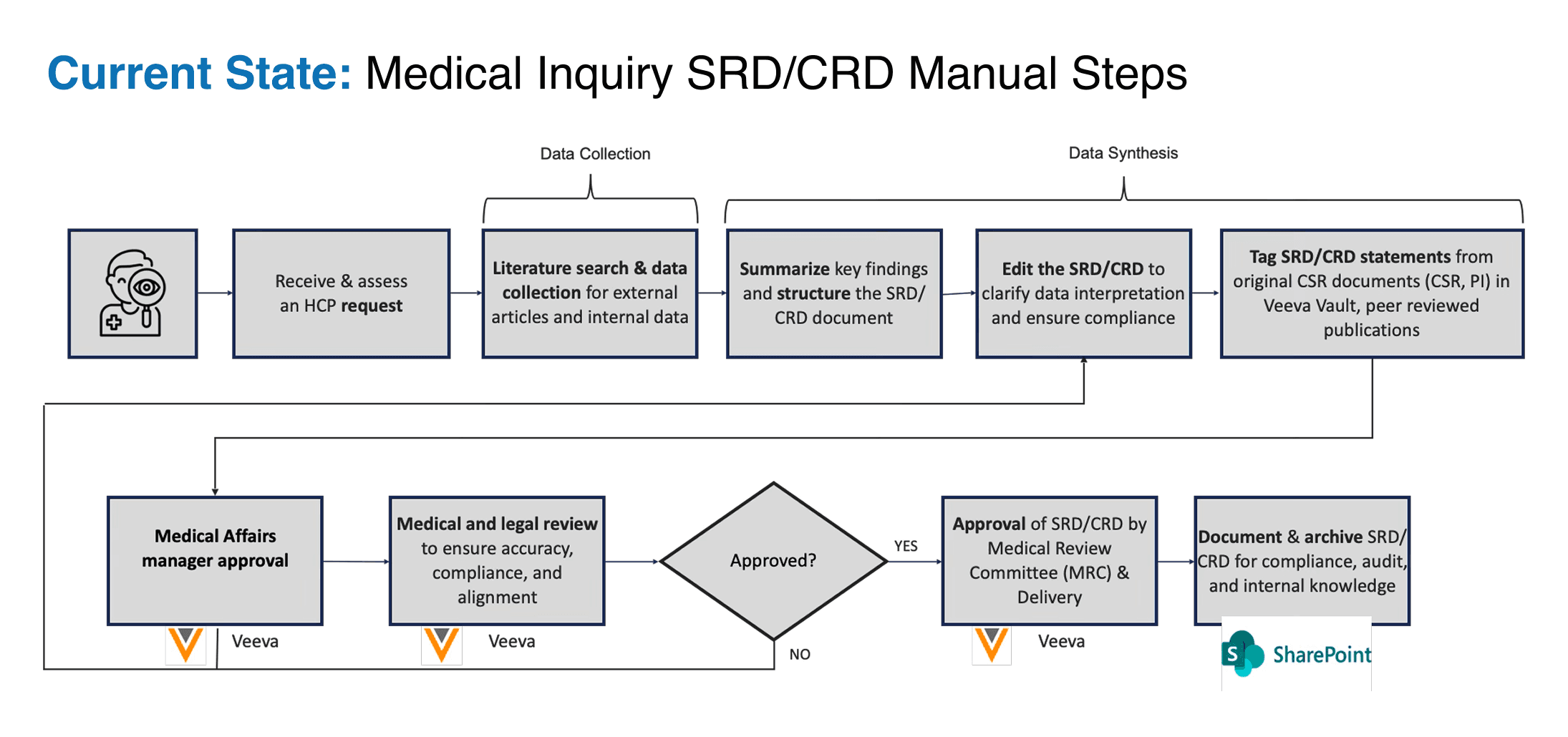
- Figure 1. The pre-AI Medical Inquiry response workflow, with manual data collection, manual synthesis, and a three-round MRC review cycle in a life sciences data management platform.
This article describes the evaluation framework we built to measure an AI-assisted authoring platform for this work, the architectural choices that shaped it, the dashboard misstep that almost defined our trajectory, and the production evidence we have gathered across approximately five months of pre-edit scoring. The platform has been in production since late 2025, and the evaluation framework has undergone three revisions and stabilized into a three-layer measurement system for a regulated medical-content workload. The work that follows is intended for engineers who have built or are considering building evaluation infrastructure for AI in regulated industries.
The Quality Problem in AI-Assisted Medical Content
Most discussions of AI in pharmaceutical document workflows begin with the time savings. They are real and substantial. What they obscure is that speed is the wrong question for any document that has to pass an MRC. A faster flawed process in regulated content is more dangerous than a slow one, because a flawed process at scale produces flawed artifacts faster than the review committee can catch them. Throughput gains that arrive before quality is measured become a liability.
The regulatory framing makes the point sharply. A medical-information response that contains a missed contraindication, a stale citation, or an interpretation that drifts beyond what the cited evidence supports is not just embarrassing. Under 21 CFR Part 11, it is a record-integrity event. Under EMA Annex 22, it is a data-quality finding. Under the FDA's recent Credibility Assessment guidance for AI-enabled regulatory submissions, the guidance raises the model risk classification of the entire system. Speed cannot answer any of those questions. Only measurement can.
The question for AI-assisted medical content is therefore not whether the model produces a draft quickly. The question is whether you can prove the draft is good. Missed studies, promotional language, stale citations, and untraceable claims are not just quality issues; they are regulatory, compliance, and clinical risks. Vendors who automate the draft but leave the committee-facing artifact untouched rarely survive a compliance audit, because the artifact the auditor reads is the document plus the chain of evidence that produced it, not the model that drafted it.
Our platform was built around continuous measurement of the artifact, with speed treated as a consequence of measurement rather than a substitute for it. That ordering is the central design decision of the system and the principal subject of this article.
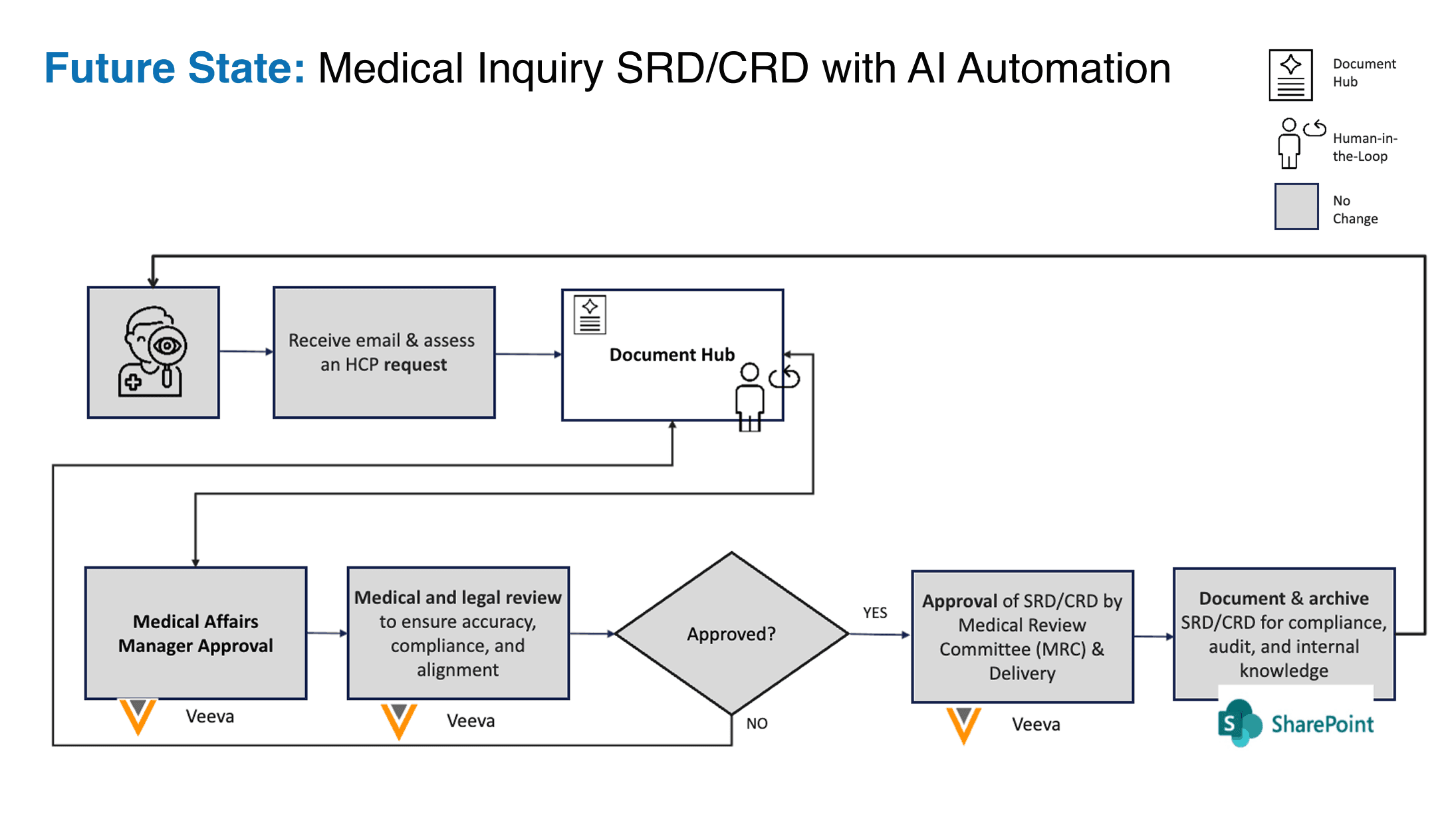
- Figure 2. The same workflow with the Document Hub in place. The bookend processes (inquiry intake and MRC approval) are deliberately unchanged from the pre-AI workflow; AI operates only inside the middle, under continuous human-in-the-loop control. The unchanged bookends are a subset of those that preserve the regulatory posture.
We built this application through our Software Factory, leveraging its agile workflow to rapidly integrate and validate the CQI evaluation framework itself, treating the rubric as a continuously refined software artifact.
The First Dashboard, and Why We Rebuilt It
The first KPI dashboard we shipped led with authoring time. The chart at the top showed median hours per document falling from a baseline of 7.5 hours toward our target of 1.5 hours, and the supporting metrics included MRC cycles per document, response time from inquiry to send, and document throughput. The numbers were real and improving. The dashboard was an honest reflection of what the engineering team had been optimizing for during the first months of the engagement. It was the wrong dashboard.
The executive sponsor, who chairs the Medical Affairs governance committee that ultimately controls the release of the platform into production use, reviewed the dashboard once and asked us to rebuild it. Her objection had nothing to do with the numbers; the numbers were unobjectionable. Her objection was to the ordering. In her words, leading with speed told the operating team that speed was what mattered. In regulated content, leading with speed is a safety inversion, because the people who read the dashboard begin to optimize for the top metric. That metric flows through quarterly planning, performance reviews, and the engineering team's prioritized work in the next sprint. A dashboard is not a passive display. It is a steering input to the organization that consumes it.
We rebuilt the dashboard around a single top-level metric, the Expert Quality Rating, with six supporting metrics arranged beneath it. The full schema, reproduced verbatim from the internal KPI document, is shown below. The top-level rating answers the question, "Is this document good enough?" The supporting metrics decompose that question into engineering-interpretable signals about approval velocity, authoring effort, retrieval coverage, recommendation fidelity, evidence overlap, and AI-to-final preservation rate.
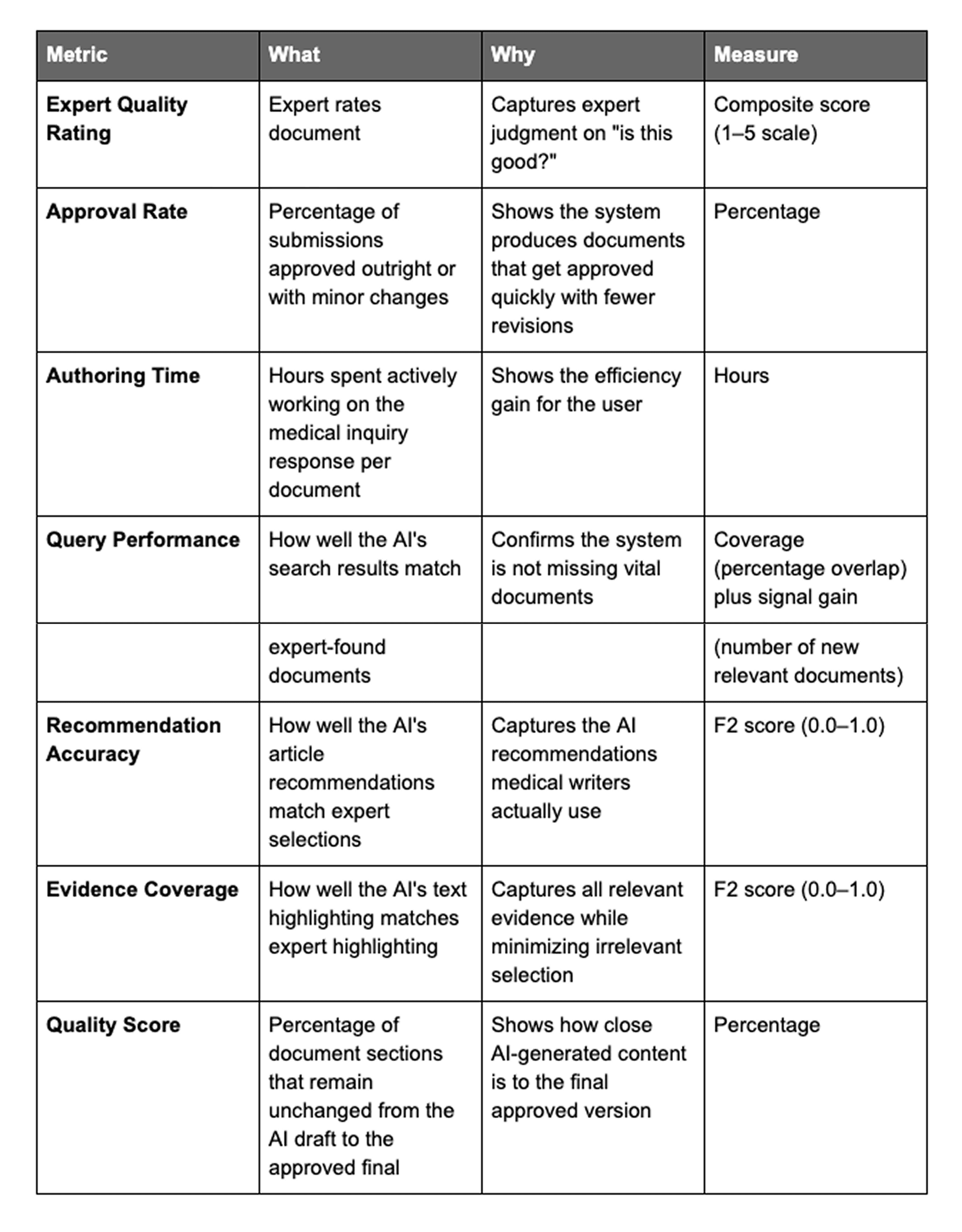
The F2 score, which appears twice in the schema, prioritizes completeness over precision: missing critical information is worse than including extra information. Speed remained on the dashboard; it moved beneath quality. The conceptual shift it produced has shaped every subsequent decision in the framework.
Architectural Principles
The eval rests on three principles, established during the rebuild and unchanged since.
Score the AI before any human edits it.
Most evaluation systems score documents after a medical writer has cleaned them up. The result is contaminated. A skilled writer rescues an unusable draft, and the post-edit score looks acceptable; a junior writer leaves the model's failures visible, and the same model appears to perform worse. In either case, the metric describes the writer-plus-AI workflow, not the AI alone. To improve the model, we needed a signal free of human correction.
The platform, therefore, captures the AI draft at the moment it leaves the model, locks the draft into an immutable evaluation snapshot, and runs the rubric against the snapshot before any human edit occurs. The medical writer then opens the document and edits freely; her edits are tracked and scored separately, generating a post-edit composite that shows how much human work was required to bring the document to approval-ready status. In practice, the post-edit work is not a single pass: each pipeline stage carries its own human-in-the-loop checkpoint, and the post-edit composite aggregates them. Obtaining a signal unpolluted by human intervention is essential for effective model refinement.
To achieve this, the platform secures an immutable evaluation snapshot of the AI draft precisely as it is generated, executing the rubric before any manual modifications occur. Once this baseline is locked, the medical writer begins editing; these subsequent changes are tracked independently to produce a post-edit composite score reflecting the effort required to reach approval-readiness. This workflow recognizes that post-edit work is iterative, with the composite score aggregating signals from multiple human-in-the-loop checkpoints across the pipeline.
We rely on the pre-edit/post-edit split as our primary decomposition, since neither metric alone fully captures the model's performance or the effectiveness of the human-AI collaboration.
Low pre-edit scores should be viewed as diagnostics rather than failures. These results pinpoint specific weaknesses, such as prompts that require revision or misclassified annotation intents, allowing the engineering team to retune the retrieval logic effectively, even when the final, human-refined document scores significantly higher. By separating these metrics within the workflow rather than reconstructing them from logs, we maintain a clear line of sight into model performance.
Pharma Risk and the Scoring Function
The selection of F2 scores for Evidence Coverage and Recommendation Accuracy is a strategic choice designed to mirror the inherent asymmetry of clinical risk. In a pharmaceutical context, failure modes are not equal. If the AI omits a pertinent clinical study, the resulting regulatory risk is severe, potentially leading to incomplete response documents or flawed treatment decisions. Conversely, if the system includes an irrelevant study, the only "cost" is a few seconds of a writer's time to reject it. Because missing information is materially more dangerous than over-inclusion, the F2 metric formalizes this imbalance. The asymmetry holds even as writers perform nuanced edits, such as adjusting scope or rephrasing, rather than simple binary selections. Consequently, the engineering team optimizes against a mathematical surface that is heavily weighted toward recall over precision. The two scores together describe both the model and the human-AI loop. Neither score alone is sufficient.
A consequence worth stating explicitly. A preedit composite of twenty percent is not a failure of the eval. It is a diagnostic, document-by-document, of where the model is weak. It tells the engineering team which prompt modules to revise, which retrieval logic to retune, and which annotation intents are most often misclassified. The same document, after the writer has finished, may score in the high nineties. We need both numbers, and we need them separated by the workflow itself, not reconstructed afterward from logs.
The scoring function must encode pharma risk.
Two of the supporting metrics, Recommendation Accuracy and Evidence Coverage, are F2 scores rather than F1. The choice is deliberate and reflects the asymmetry of clinical risk.
Consider the failure modes. If the AI fails to return a relevant clinical study, the medical writer must manually catch the omission. An omission is a regulatory event. The response document may be incomplete; the MRC may flag it on review; and, at the limit, a treatment decision could be made based on a response that omits material evidence. If the AI returns one extra study that is not strictly relevant, the medical writer rejects it during annotation, and the cost is a few seconds of her attention. The two errors are not symmetric. Missing a relevant study is materially worse than including an extra one. In practice, the medical writer makes many incremental modifications rather than clean accept/reject decisions on each AI suggestion: partial inclusion, scope adjustments, and rephrased framings. The binary framing above is a simplification that makes the F2 choice legible. The under-retrieval/over-retrieval asymmetry holds across the full edit spectrum, as F2 captures.
The F2 metric formalizes this asymmetry:
The optimization surface the engineering team works against is mathematically weighted to prioritize recall over precision.
By weighting recall four times as heavily as precision in the denominator, F2 ensures that the optimization surface the engineering team works against matches the operating risk the medical writer experiences. We considered F1 and we considered precision-only; both encode the wrong bias for regulated content. The KPI doc footnote that has survived three rubric revisions states the principle in nine words: missing critical information is worse than including extra information. That sentence is the entire pharma-evaluation thesis, and we recommend it to any team that is choosing scoring functions for AI in a regulated workflow.
Source-grounding by architecture, not by policy
The third principle is that every claim in every generated document must be traceable back to an approved source. This is not a stylistic guideline. It is a hard architectural constraint, enforced at generation time rather than at review time. If a generated sentence cannot be tied to a specific passage in a specific version of a specific source document, the system rejects the output and re-prompts the language model.
Traceability is enforced at the sentence level. Every query-relevant or significant sentence the AI emits carries a structured citation anchor pointing to a specific text bounding box in the source PDF. The chain of custody runs from each generated sentence to its citation, to the annotated passage, to the specific pixel region in the source document. We also block plagiarism architecturally: any verbatim copying of more than a configurable number of consecutive words triggers a re-prompt, with the prompt augmented to require original phrasing. This threshold was chosen empirically and is regularly tested, as shorter thresholds produced false positives with common medical phrasing (drug names, anatomical terms, standard dosing language), while longer thresholds missed lazy paraphrasing.
The result is that the eval is not asking the rubric to detect ungrounded claims after the fact. Ungrounded claims are blocked at generation. The rubric measures the quality of what survives the architectural filter, which is a much smaller and more interesting space.
Evaluation Framework
With those principles fixed, the eval is built as three layers operating in parallel against the same locked pre-edit draft. Each layer answers a different question, and the three together produce a Composite Quality Index (CQI) on a zero-to-one-hundred scale that encodes pharma risk by weighting safety-critical omissions more heavily than inclusion errors.
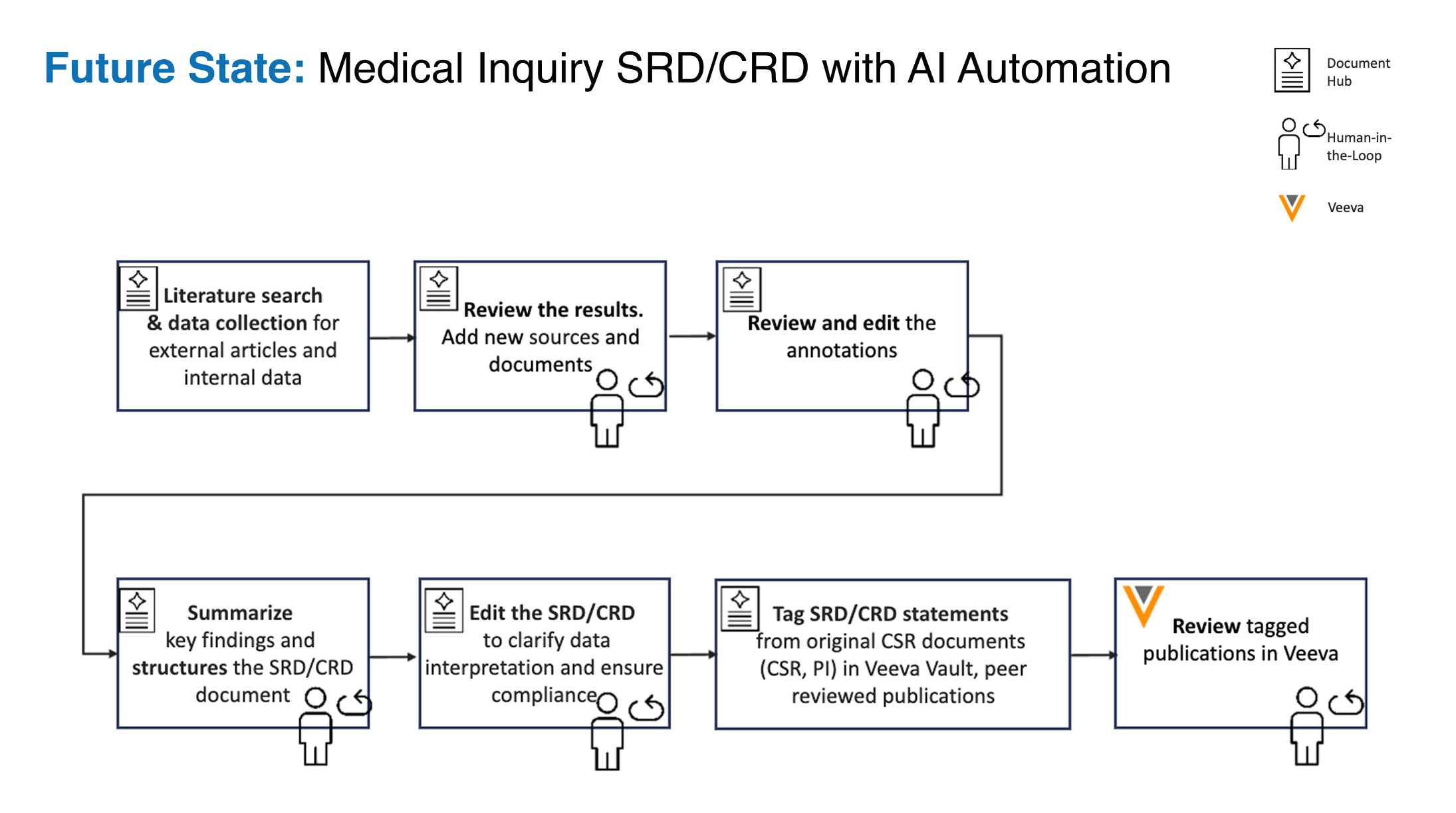
- Figure 3. The Document Hub itself, with the six gated authoring stages and a human-in-the-loop checkpoint on each. Layer 1 of the evaluation framework consists of a structured assessment form that the medical writer completes for each document at each stage.
Layer 1: The six-stage assessment form
The medical writer completes a structured assessment form for each document, with one section for each stage of the authoring workflow. The six stages are: query formation, literature search, annotation, draft generation, review, and document management upload. For each stage, the writer rates the AI's performance on the criteria specific to that stage: the precision of the Boolean query for stage one; the relevance of the search results for stage two; the accuracy of the highlighted passages and the appropriateness of the assigned annotation intent for stage three; the fidelity and tone of the generated draft for stage four; and so on. The form takes the writer approximately seven minutes per document and produces a stage-resolved view of where the AI is performing well and where it is weak.
The form is the data source from which everything else is calibrated. It is the only layer that incorporates direct human judgment on every document. The other two layers are validated against it.
Layer 2: Two independent rubrics in parallel
Above the assessment form, two independent rubrics run in parallel against the locked draft. The first is an eight-criterion rubric oriented toward completeness, evidence quality, and citation integrity. The second is a ten-criterion rubric oriented toward clinical tone, regulatory appropriateness, and submission readiness. This dual-rubric design ensures comprehensive coverage against the asymmetric risk set inherent in medical document authoring; a quality gap that one rubric is not equipped to catch is caught by the other.
The dual-rubric design replaced an earlier single eight-criterion rubric we had used through January 2026. The single rubric had two failure modes that we documented during routine use. First, it was applied after the writer had edited the document, which produced the contamination problem described in section 4.1. Second, even after we moved scoring to the pre-edit draft, a single rubric could not catch gaps that fell outside its criteria; a rubric for citation integrity, by design, is silent on tone. The dual rubric, with deliberate overlap, was the simplest mitigation we could verify that produced more useful scoring.
Layer 3: An LLM-as-Judge against a Golden Dataset
The third layer is an automated evaluator built on a language-model judge, calibrated against a Golden Dataset of human-scored production documents. The Golden Dataset is the corpus that the engineering team uses to verify that automated scoring aligns with human reviewer judgment. We re-validate the judge against the Golden Dataset on a quarterly cadence; if the agreement between the judge and the human reviewers drifts below a threshold, we recalibrate before the next production cycle.
The judge layer is what makes per-document scoring tractable. Layer 1 requires a medical writer's time, which is the most expensive input the system consumes. Layers 2 and 3 are automatic. By calibrating the judge against the human-scored Golden Dataset, we get a measurement signal that approximates human judgment at machine throughput, with quarterly human verification.
The Composite Quality Index aggregates the three layers. It is the headline number on the dashboard, on the CEO update deck, and in this article.
Production Trajectory
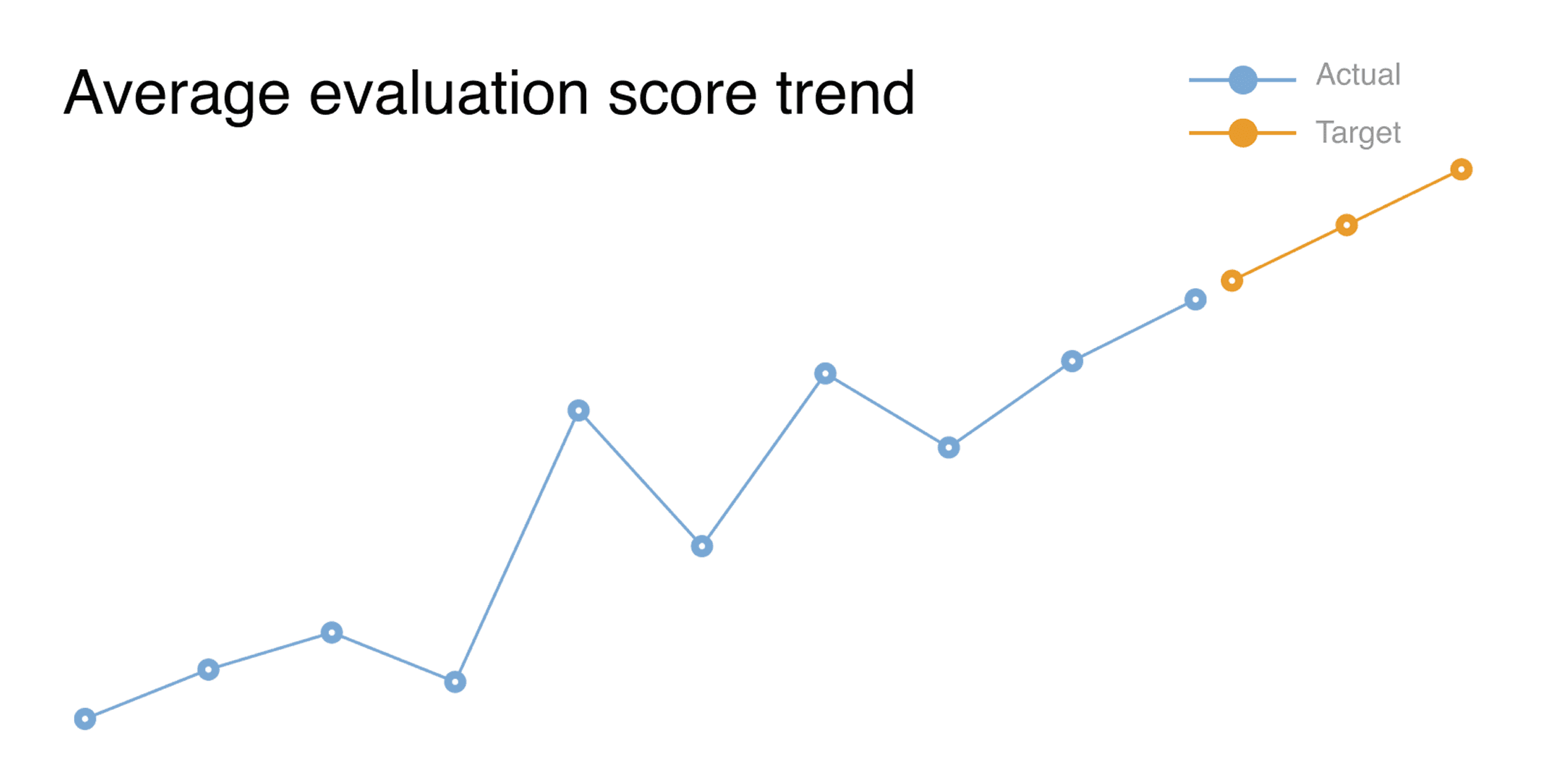
The score went up over the first few months, but not in a straight line. After the platform stabilized, we switched from monthly readings to weekly ones. The weekly view showed something the monthly view would have missed. Two things stood out.
The first is a dip in the score that showed up right after we hit a milestone we called "Stability and Feature Complete." That looks like the wrong direction: the platform was supposed to score better after stability, not worse. The cause was specific. Before that milestone, the end-to-end pipeline often broke before documents reached the later parts of the rubric. When the pipeline ran through to completion, the rubric began scoring parts of the AI's output that earlier failures had hidden. The dip was not the model getting worse. It was the rubric, seeing more of the model than it had before.
A synthetic benchmark runs the model on a fixed test set and produces a consistent score. A production eval runs the model within a real workflow, and the workflow's failures determine how much of the model is measured. When you fix the upstream failures, the eval starts measuring more. A dip after a big milestone is, in our experience, what a production eval looks like when it is working. We expect it now, and we treat it as a sign that the earlier numbers were only telling part of the story.
The second thing worth flagging is the sample size. We have measured a set of documents in a single therapeutic area. We have not yet measured how scores compare across customers, therapeutic areas, or over multiple quarters. These are real limits, and the trend is suggestive, not proven. A larger, more varied sample, which we would get by running the platform with multiple customers, is the next big step for the framework.
The Engineering Cost of a Production Evaluation
A production evaluation has fixed operational costs that a research benchmark does not, and those costs are what make it trustworthy.
Every AI operation on the platform produces an LLM Decision Record: a structured log that captures the session identifier, the pipeline stage, the model identifier, the temperature, the timestamps, the input and output token counts, the input and output summaries, the reasoning trace (where available), and a confidence score. The decision chain is retrievable and filterable, the scoring layer can re-attach the record to the eval result, and the records are append-only to support the audit trail required under 21 CFR Part 11. The records run to several megabytes of structured data per document. Storage is not the constraint; query performance and dashboard latency are, and we have invested in both.
The Golden Dataset is the second fixed cost. Building it required senior medical writers to score approximately 50 historical, pre-AI production documents against the dual rubric, establishing a human-judgment baseline against which the LLM-as-Judge could be calibrated. We adjudicate disagreements between writers, and a quarterly recalibration cadence pulls the same writers off authoring work for several days per quarter. The Golden Dataset baseline is distinct from the post-relaunch cohort of approximately twenty-three live, AI-generated documents tracked in section 6: the first is historical reference material used to calibrate the judge; the second is the production trajectory we are measuring. This is the single most expensive line item in the eval and the one we have most often been tempted to reduce. We have not reduced it, because every time we have considered loosening the recalibration cadence, we have remembered that the Layer 3 judge is the bottleneck signal for the dashboard. The judge is only as trustworthy as the Golden Dataset it is calibrated against.
The third cost is the seven-minute per-document burden on medical writers to complete the Layer 1 assessment form. We have considered automating the form. We have not done so because the writer's per-stage rating is the only direct human-judgment signal the eval receives for every document, and removing it would convert the framework from a measurement system into an unverified estimator. We pay the seven minutes because there is no substitute for it.
We mention these costs because the most common failure mode of vendor evaluation frameworks is to promise that no operational burden falls on the customer team. That promise is incompatible with a measurement signal that survives audit. The eval that costs nothing to run is, in our experience, the eval that cannot be defended in a regulatory inspection.
Compliance Posture as an Architectural Argument
Our compliance posture is an architectural outcome: the LLM Decision Record substrate serves as the primary evidence base for GAMP 5 qualification, ensuring every model decision is traceable by design.
Treating the platform as a GAMP 5 Category 5 system, we use the locked pre-edit snapshots and decision logs to provide an automated evidence base for qualification. This architecture ensures that traceability between requirements and behavior is a technical guarantee rather than a manual documentation task, supporting our V-Model execution through June 2026.
The system encodes ALCOA+ principles by using append-only versioning and immutable JSON logs. By embedding data integrity into the infrastructure via the LLM Decision Recording substrate, we satisfy 21 CFR Part 11 requirements through technical controls that prevent deviations from being hidden.
We completed an FDA-style Credibility Assessment for AI-enabled regulatory use and had it reviewed by external FDA regulatory counsel in early 2026. The Context of Use is defined for each of eight LLM output categories, with temperature controls from 0.0 to 0.3 selected by category rather than uniformly. Every LLM Decision Record captures the model identifier and the temperature at the time of inference. Hence, a model change is detectable, and a deviation from the approved Context of Use is auditable.
Our FDA-style Credibility Assessment enforces specific temperature ranges (0.0 to 0.3) for each of eight output categories. Every record captures the exact inference parameters, making model drift or unauthorized deviations from the Context of Use immediately auditable.
Changes to AI components require unanimous clearance from all seven risk functions. This governance model has successfully processed a dozen production updates since January without a single veto, guided by a 33-point risk taxonomy aligned with the EU AI Act.
The 33-point enterprise AI risk taxonomy underlying our governance aligns with the EU AI Act risk categories and is reviewed quarterly to account for the evolving regulatory landscape. Twenty-four of the thirty-three risks are classified as in-scope for the platform, distributed across six high-, four medium-, and fourteen low-impact categories.
Limitations and Open Problems
The framework has known limitations, and the engineering team is candid about them with the customer, with prospective customers, and (in this article) with the broader engineering community.
The most important limitation is the eval cohort size. Twenty-three documents across a single therapeutic area constitute a small base for strong claims about generalizability. We treat the production numbers in section 6 as indicative, rather than authoritative, and explicitly note this gap. A larger, more diverse cohort, ideally enabled by a multi-tenant deployment, is the next major milestone.
The second limitation is cross-tenant calibration. Every customer onboards with their own rubric calibration tuned to their therapeutic areas, their literature mix, and their MRC reviewing conventions. We do not yet have a methodology for comparing CQI scores across customers that respects these differences without flattening them. Cross-tenant comparability is more.
Interesting benchmark question; it is also the one we have not solved.
The third is mitigating judge drift. LLM-as-Judge approaches have well-documented reliability issues, including positional bias and sensitivity to prompt phrasing. While our quarterly Golden Dataset recalibration cadence mitigates these issues, it does not eliminate them. Changes in document types, specific source data sets, or writer behavior can introduce drift. We have considered increasing the recalibration frequency to monthly, but the operational costs are significant, and the trade-off remains unresolved.
The fourth is the cross-customer Golden Dataset problem. The Golden Dataset is, by construction, drawn from a customer's production documents. A second customer cannot benefit from the first customer's Golden Dataset under data-handling terms. However, the framework would be more powerful, and cross-tenant calibration (Limitation 2) would be tractable if a privacy-preserving methodology for extending the Golden Dataset across customers existed. We do not yet have one.
Conclusion
Fourteen months in production distilled these three non-negotiable principles for regulated AI. Quality must anchor speed, not the other way around. In regulated document authoring, a faster flawed process is a liability. Leading with speed metrics is a safety inversion; leading with quality ensures the system optimizes for compliance and clinical accuracy. The scoring function must encode asymmetric domain risk. Generic metrics fail regulated contexts. Clinical risk is asymmetric: a missed contraindication is a safety event; an extra search result costs a few seconds of human attention. Therefore, the optimization math (F2 scoring) must align with the operational stakes. Measure the model, not the cleanup. Post-edit scores are contaminated by human skill. To identify and repair model weaknesses, we enforce scoring on the raw AI output (pre-edit snapshot). This architectural isolation converts evaluation from a vanity metric into a diagnostic tool for engineering.
If these problems read like the kind of engineering you want to be doing, we are hiring. See https://www.8090.ai/careers.

-1.png&w=3840&q=75&dpl=dpl_8vh3BMyGSxqCUUkQJJV3p98o2YHQ)
-2.png&w=3840&q=75&dpl=dpl_8vh3BMyGSxqCUUkQJJV3p98o2YHQ)